10X Genomics
I worked at 10X Genomics between October 2014, when they were still stealth, and May 2017. My projects were primarily on the DNA sequencing platform using molecular barcodes attached in emulsion to add long range genetic information to short read nextgen sequencing.
OVERVIEW
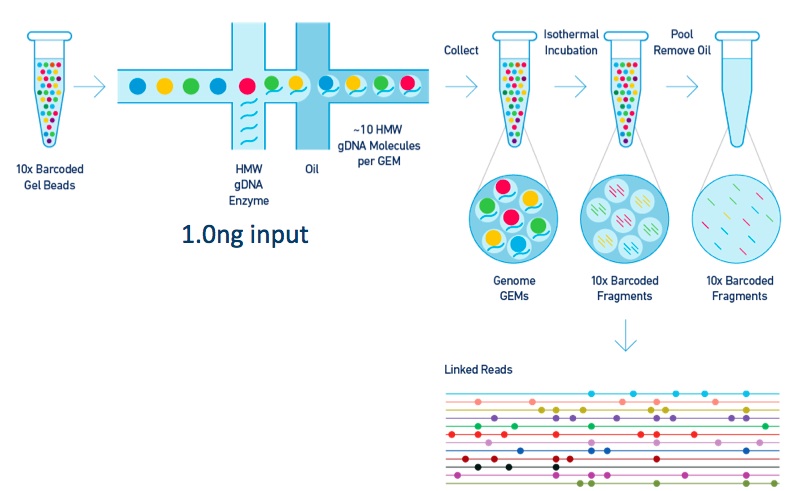
The 10X platform starts with high molecular weight DNA input into a microfluidic system which partitions those long DNA molecules into “GEMs” (Gel bead in emulsion) with oil surrounding an aqueous solution containing the DNA and reagents surrounding a gel bead containing millions of copies of the same barcode DNA sequence. Each different gel bead has a different barcode DNA oligo with high probability. Each GEM gets roughly 10 long DNA molecules. So when you map the reads from a single barcodes, they cluster into a few small regions of the genome associated with their molecule of origin. This long range information can then be used to map into repeat regions of the genome, phase haplotypes, and call structural variation.
This can be done for either whole genome or targeted sequencing while retaining the linked read long range information.
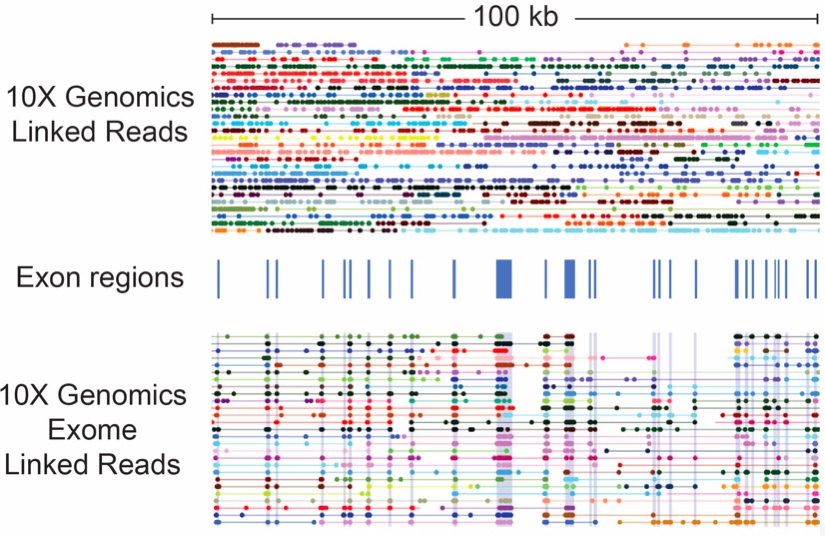
MAPPING REPEAT REGIONS OF THE GENOME
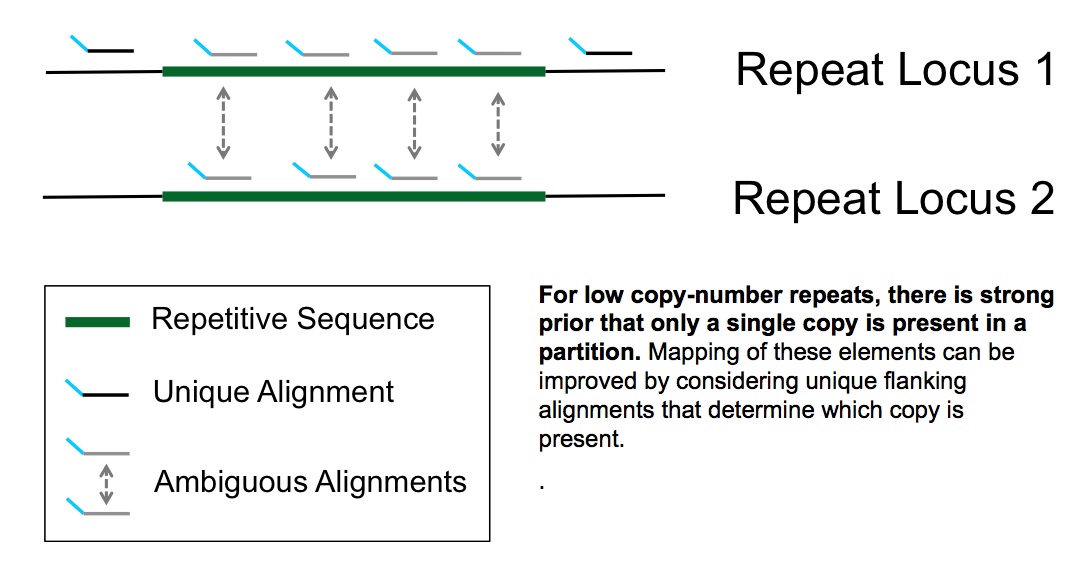 This long range genetic information allows us to map into repeat regions
of the genome using either flanking unique sequence as in the case or short exact repeats or rare interspersed differences typical of longer
segmental duplications. Segmental duplications are increasingly being recognized as the source of new gene creation as well as gene dosing variation.
You can read more about this method on my white paper. I was lead on this project and presented my findings
at Genome Informatics in September 2016. My poster is here.
This long range genetic information allows us to map into repeat regions
of the genome using either flanking unique sequence as in the case or short exact repeats or rare interspersed differences typical of longer
segmental duplications. Segmental duplications are increasingly being recognized as the source of new gene creation as well as gene dosing variation.
You can read more about this method on my white paper. I was lead on this project and presented my findings
at Genome Informatics in September 2016. My poster is here.
PHASING LONG MOLECULES
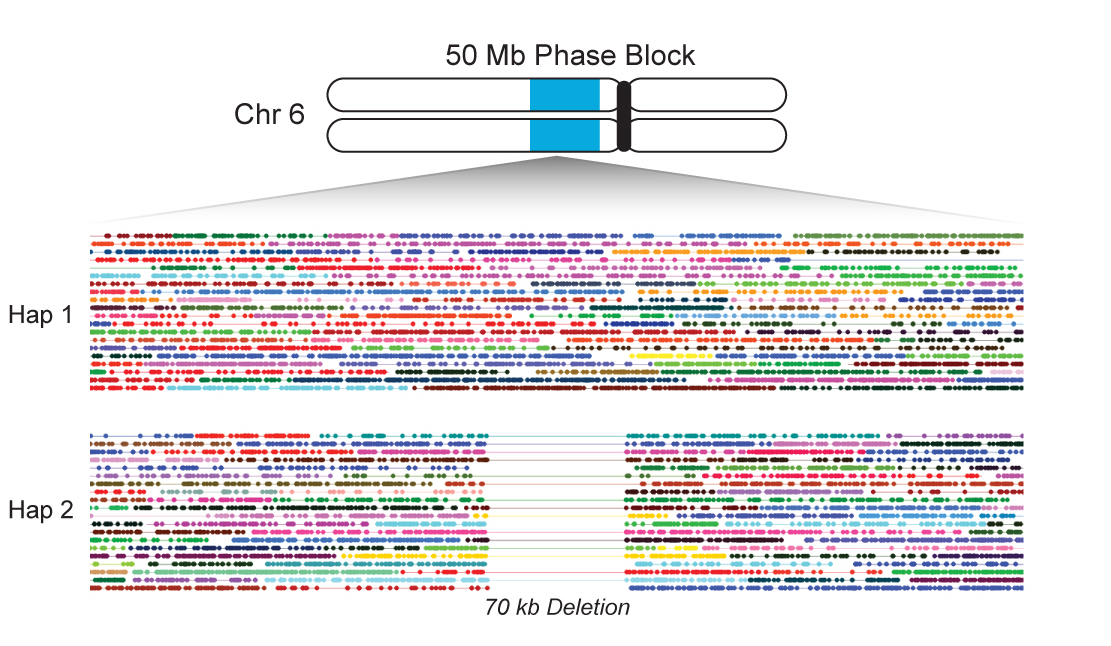
The 10X platform can also be used for haplotype phasing. Because there are so many partitions/barcodes and each barcode has so few DNA molecules, the chance that a barcode has 2 molecules from the same locus in the genome but opposite haplotypes is vanishingly small. Based on this, for each cluster of reads on the genome from one barcode, the chances that all of those reads come from the same haplotype is exceedingly high. We can use this long range information to link alleles from multiple heterozygous variants into haplotypes. Then more molecules can link those alleles to even more alleles and proceeding this way we can create phase blocks that span megabases or even whole chromosome arms.
I contributed to the phasing algorithm and extended it from phasing variants to phasing molecules and all of their reads. With this information it makes normally difficult to call variant types like heterozygous deletions much easier. I also designed and implemented an algorithm to use the phasing to filter false positives that do not segregate on haplotype lines. In this same algorithm I can correct genotypes of variants that were falsely called heterozygous which were in reality homozygous. The details of this algorithm can be found on our phasing white paper. And the linked read papers can be found here and here.